
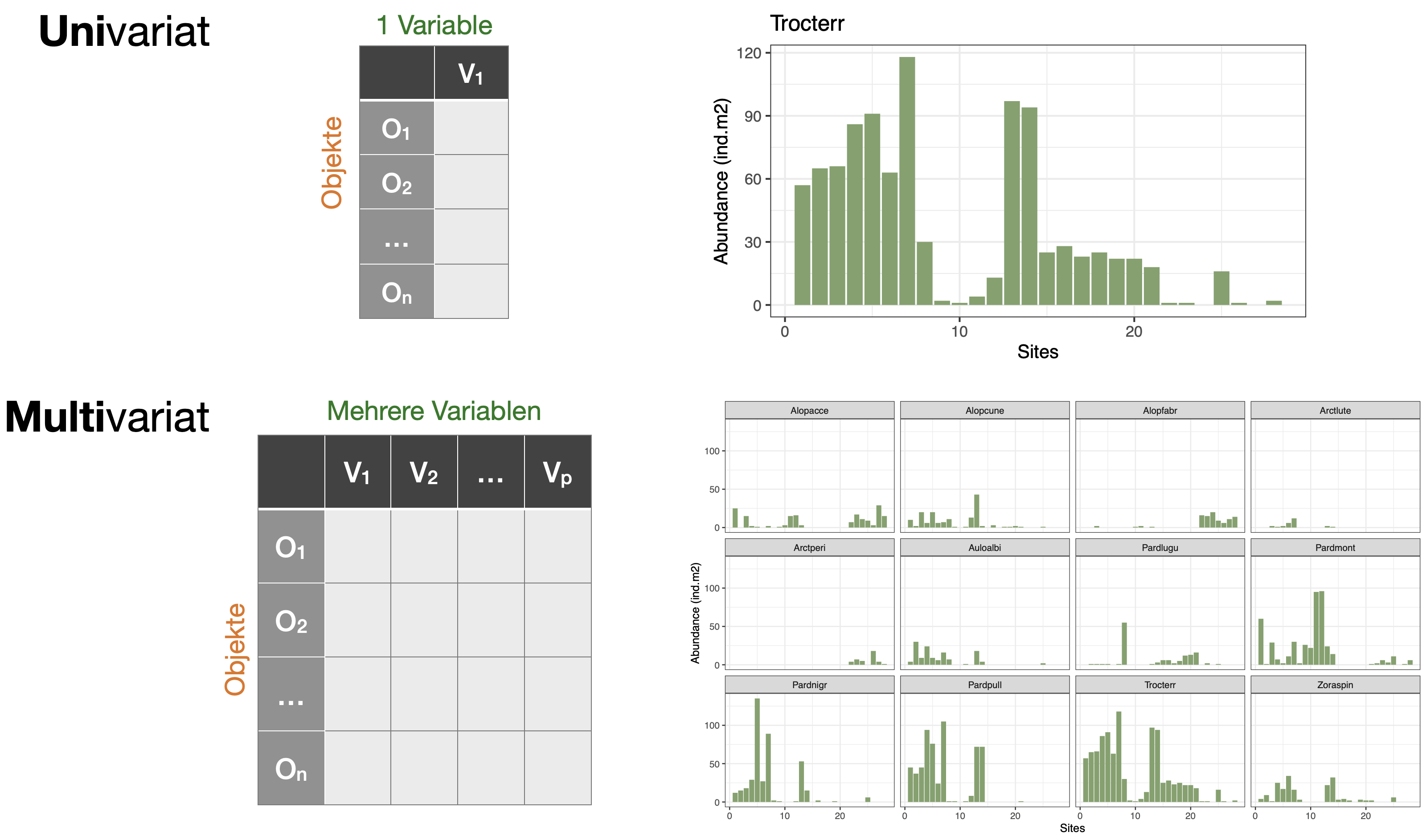


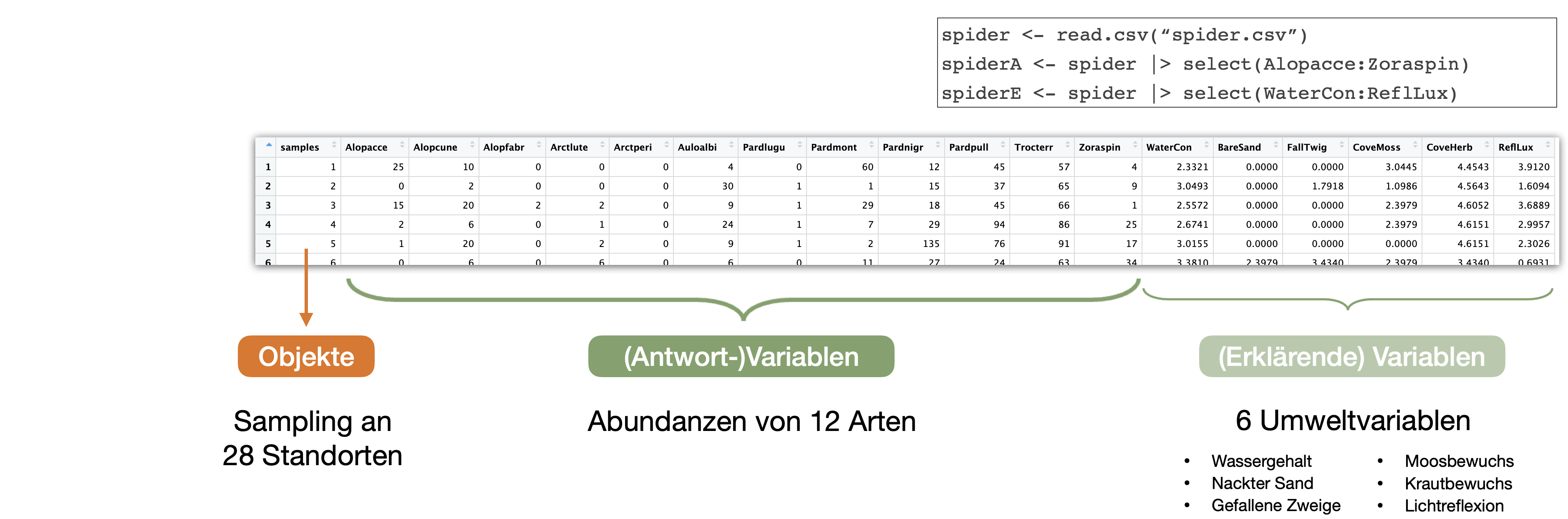
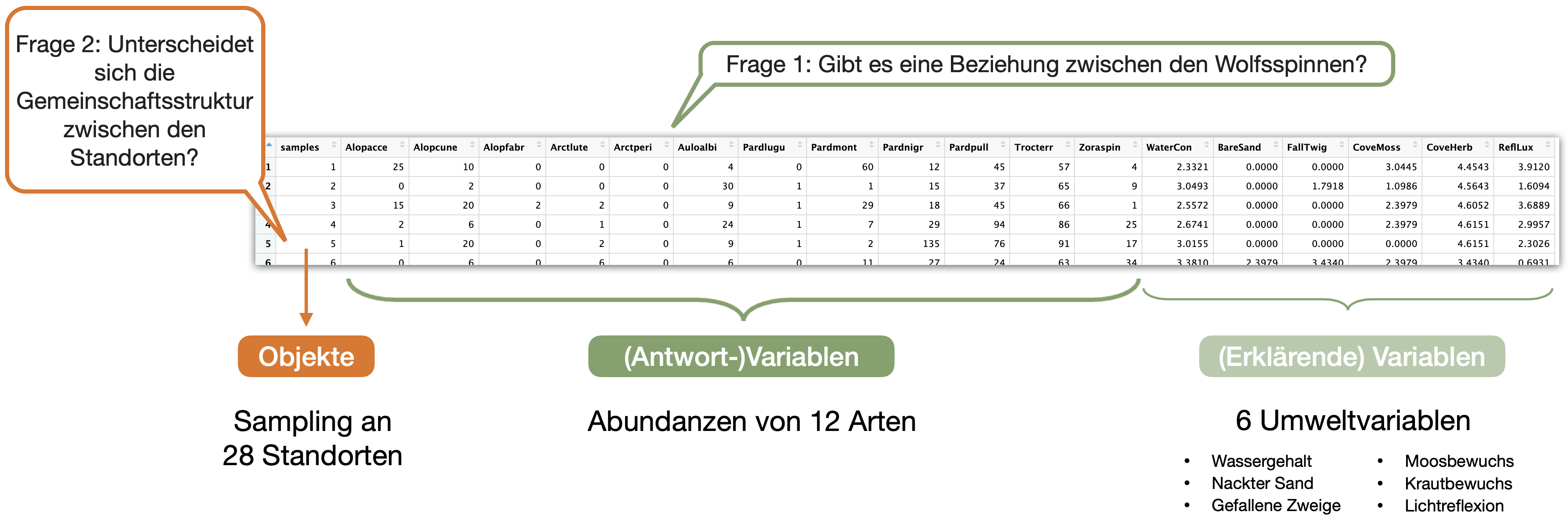

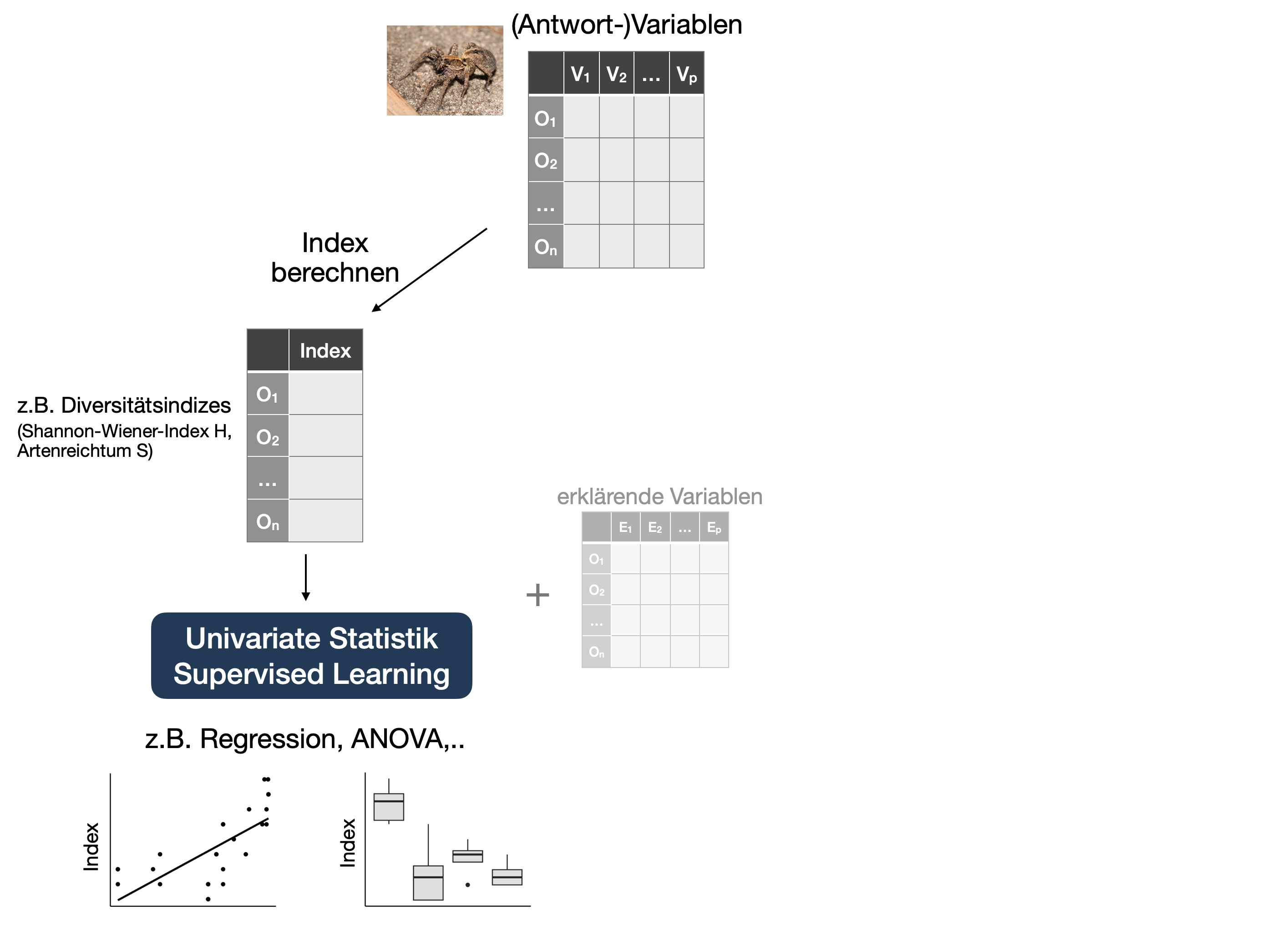
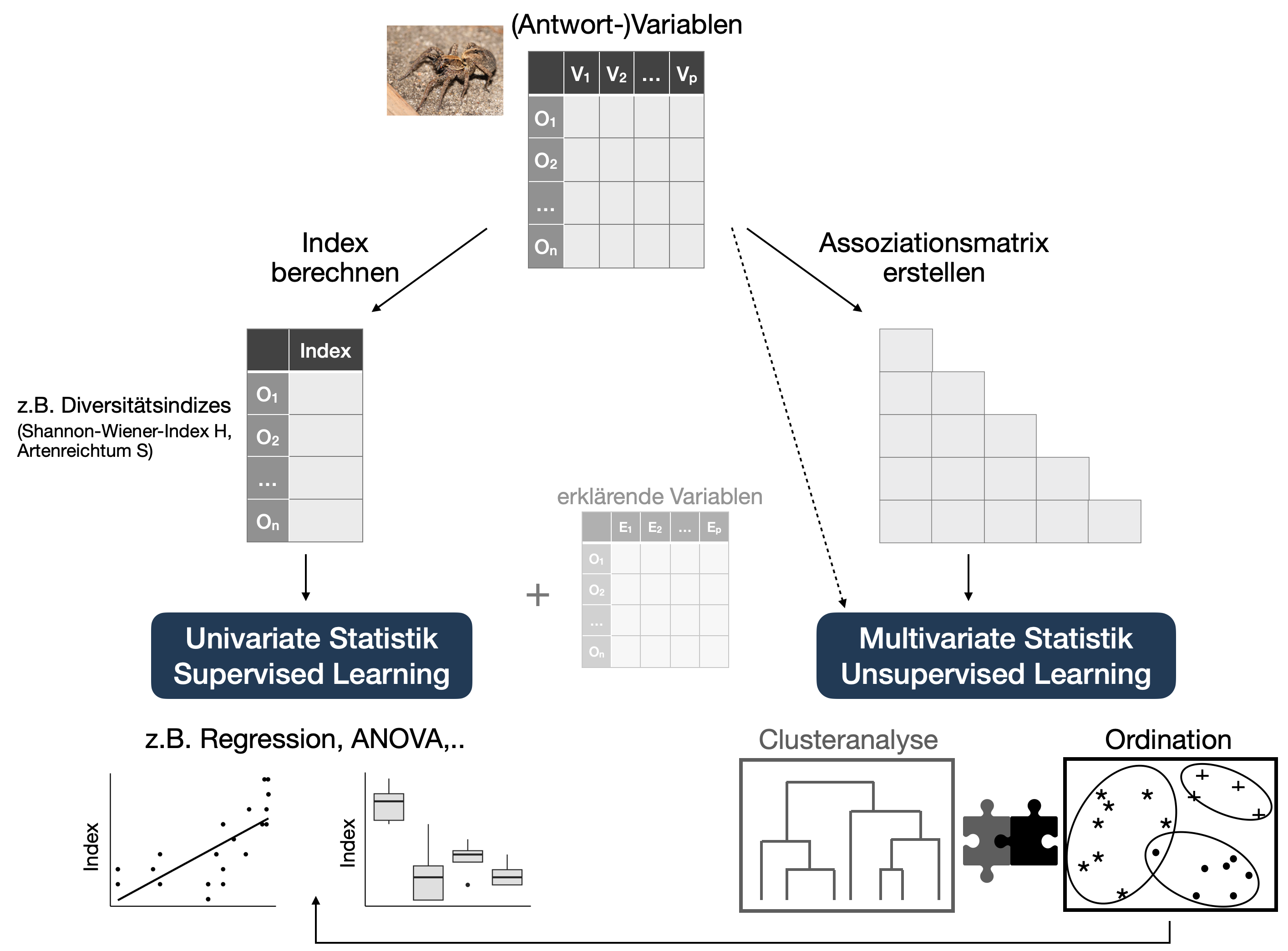
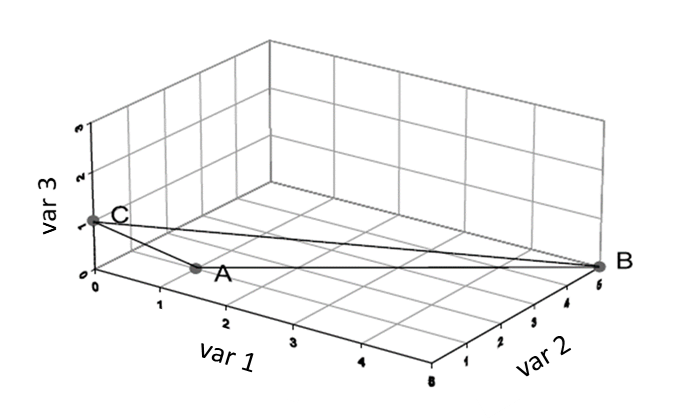
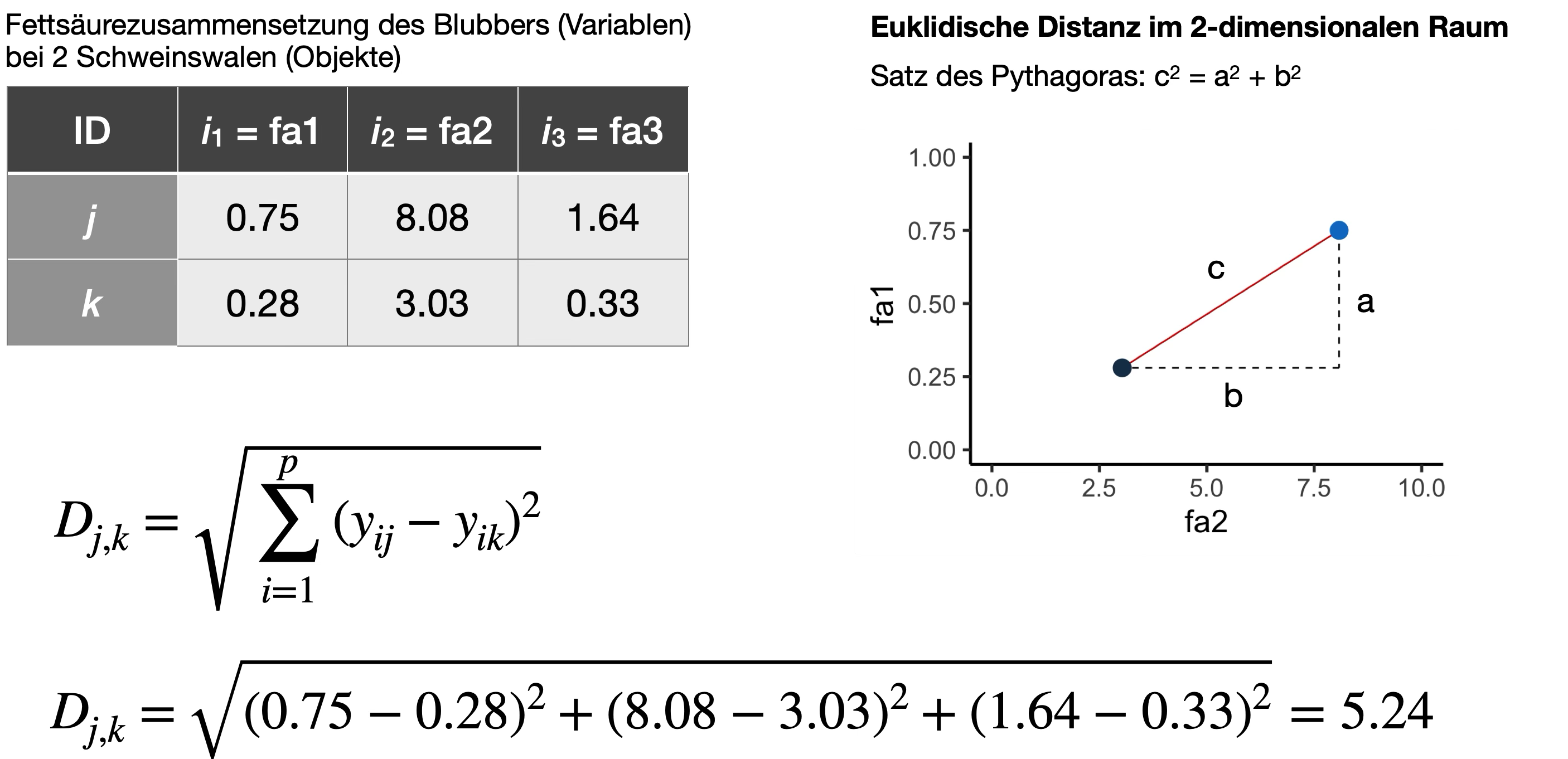
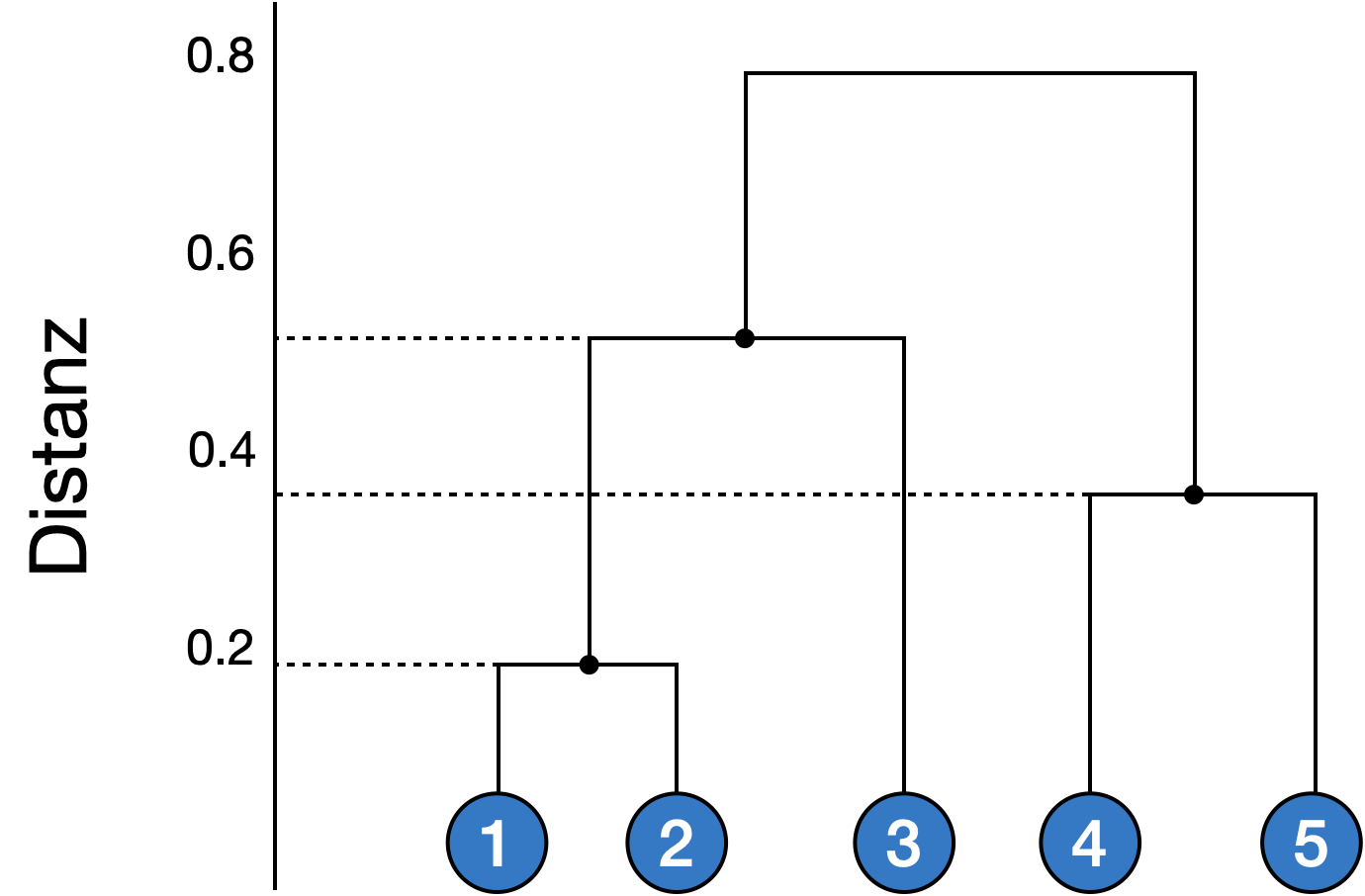
Artenreichtum S und Shannon Index H mit 'vegan' berechnen:
samples S H
1 13 11 1.8742271
2 3 11 1.8732193
3 7 10 1.7221199
4 4 10 1.6520490
5 5 10 1.5403056
6 25 9 1.9276654
7 14 9 1.5284480
8 6 8 1.7591828
9 1 8 1.7309509
10 2 8 1.5204418
11 8 8 1.3896573
12 11 7 0.7440770
13 23 6 1.4848760
14 21 6 1.1565256
15 12 6 1.1410240
16 9 6 0.7752108
17 28 5 1.2769439
18 22 5 1.2373886
19 16 5 1.1035352
20 19 5 1.0708014
21 24 4 1.1601124
22 20 4 0.9938215
23 26 4 0.9724515
24 27 4 0.9272491
25 10 4 0.6551416
26 15 3 0.7464108
27 18 3 0.5785328
28 17 3 0.5221718


















.jpg){kind=link}
{kind=link}