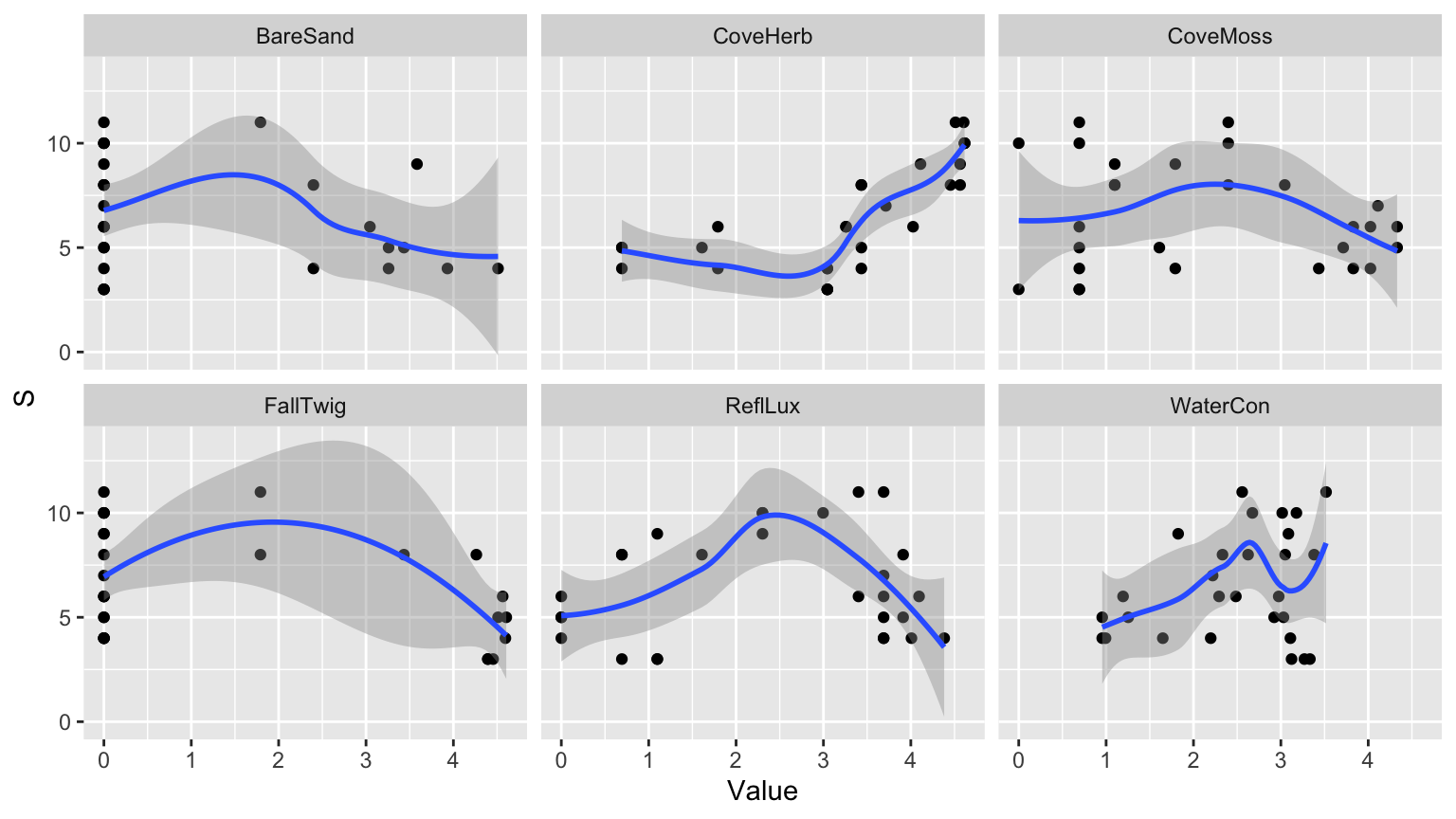
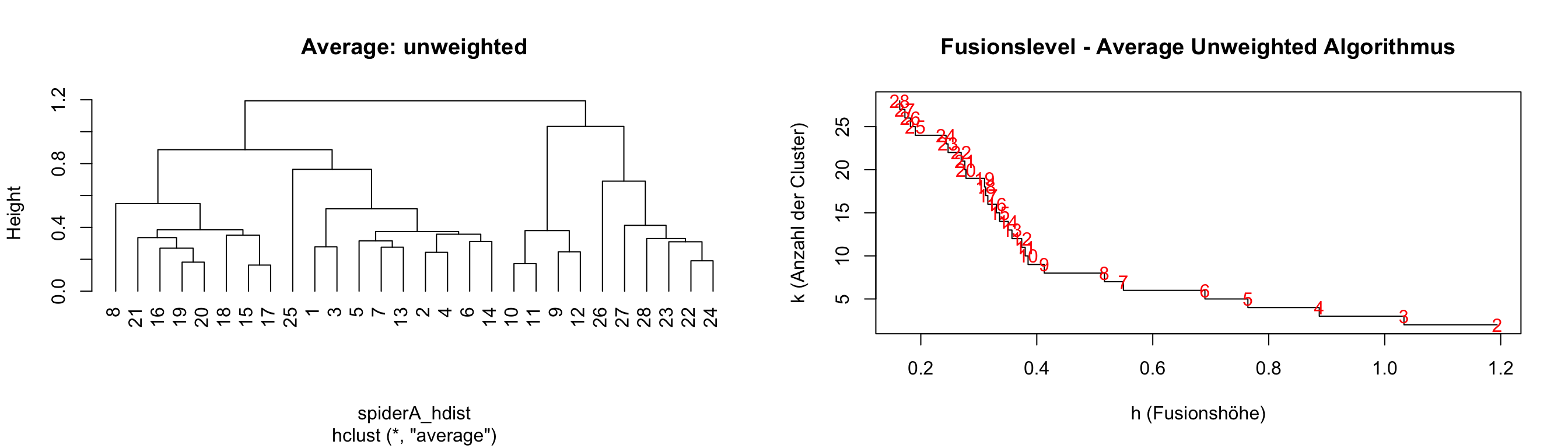
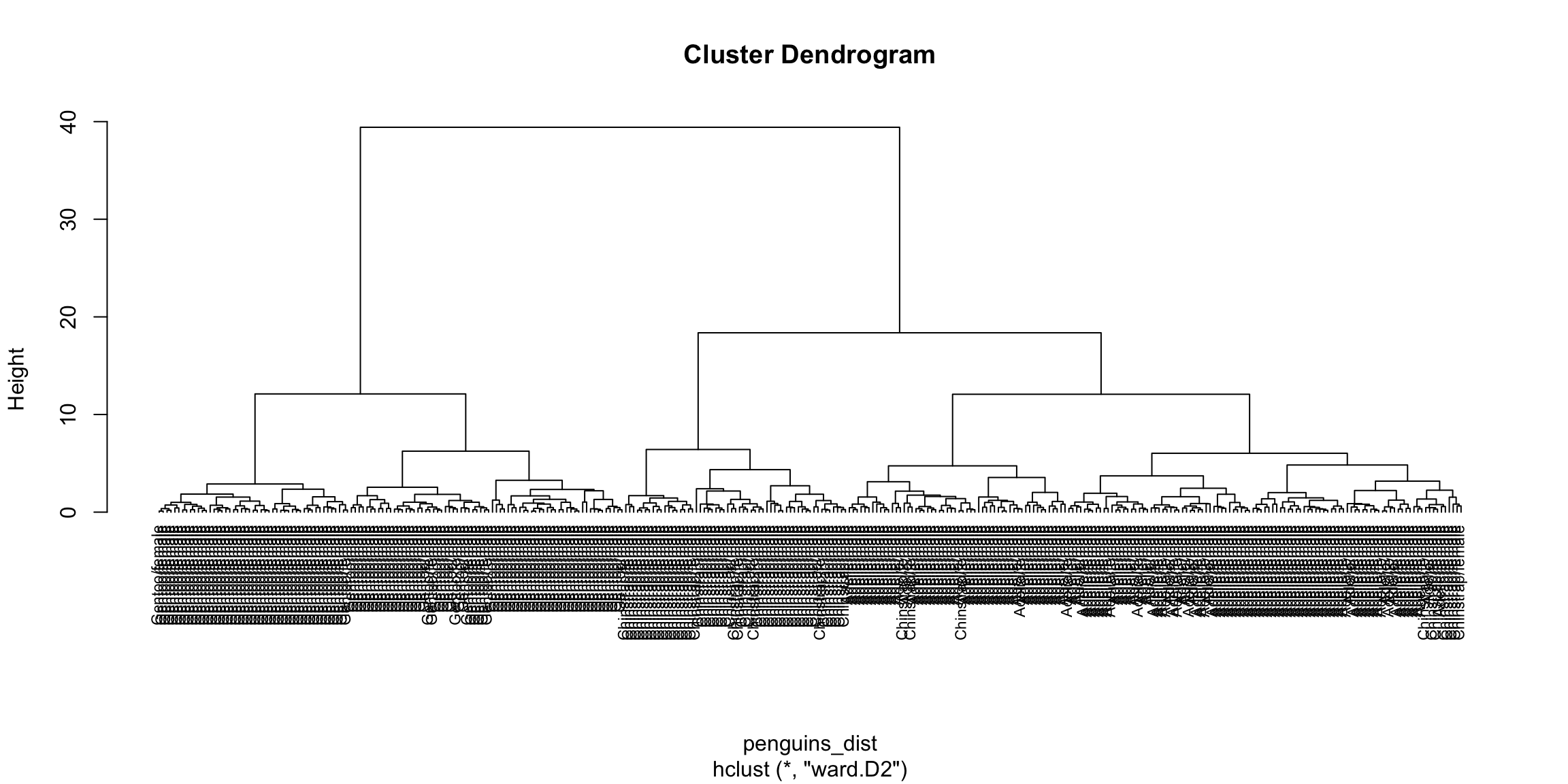
par(mfrow = c(1,2))
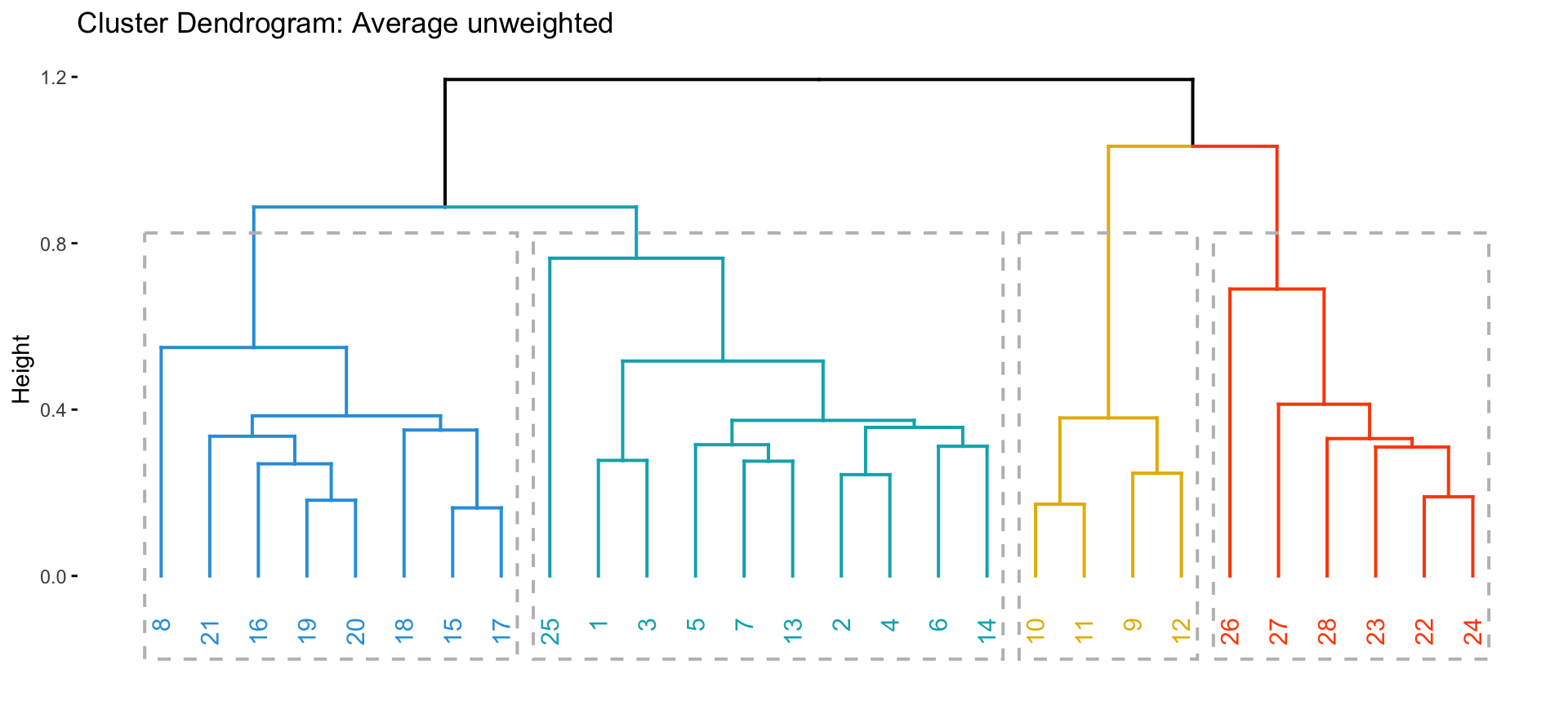
plot(hc_ave, main = "Average: unweighted", hang = -1)
# Plot der Fusionshöhen und Clusteranzahlen
plot(hc_ave$height,
nrow(spiderA_hell):2, # Anzahl der Cluster von n bis 2
type = "S", # "S" für Step-Plot
main = "Fusionslevel - Average Unweighted Algorithmus",
ylab = "k (Anzahl der Cluster)",
xlab = "h (Fusionshöhe)"
)
# Text in rot für die Clusteranzahlen
text(hc_ave$height,
nrow(spiderA_hell):2, # Clusteranzahl entlang der Y-Achse
labels = nrow(spiderA_hell):2, # Beschriftung der Clusteranzahl
col = "red"
)

















.jpg){kind=link}
{kind=link}